选择压力分析基本原理
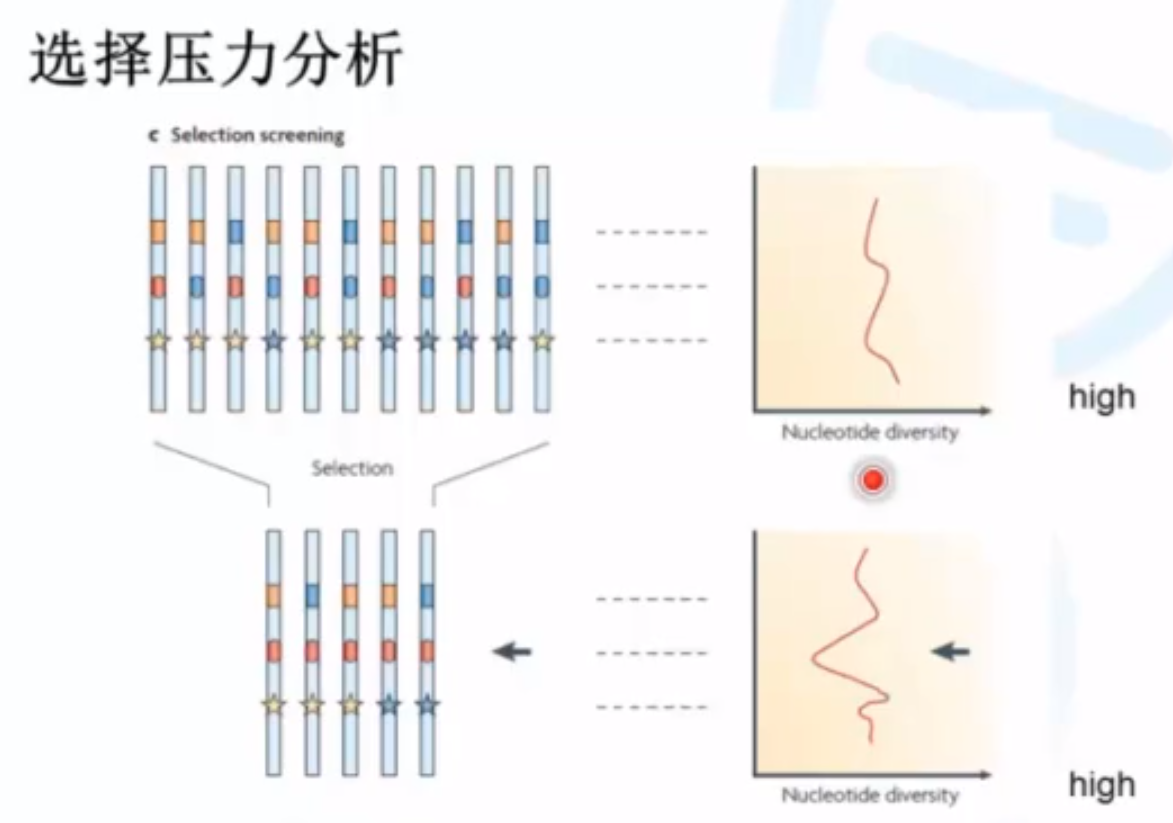
原始群体中,遗传多样性是十分高的,整个序列的核酸diversity都高。而在受到选择之后,diversity会发生波动。核酸多样性下降 可能就是由于under selection导致的。
在演化/驯化过程中,如果某一基因X占优势,即X的基因型占据主导地位,则基因X所在区域的杂合率/多样性会显著下降。本质就是 比较基因组不同区域多样性(杂合率)的变化
- 群体遗传关心的问题:
- 遗传结构(phylogeny+structure)
- 基因组上受选择区域:群体水平基因组不同位置的区域遗传多样性变化的规律(例如:Pi、Tajima’s D, Fst)
- 变异类型:
- 中性突变(同义、相同类型的氨基酸、不影响环境适应性):平衡选择,这种基因型频率是大致恒定的
- 有利突变(正选择):选择扫荡(Selective sweep),与有利突变的中性突变的频率会显著提升
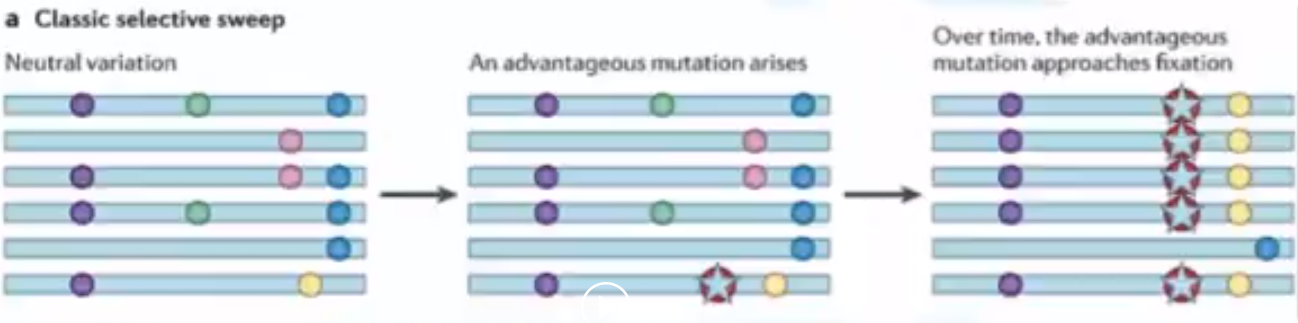
- 有害突变(负选择):背景选择(negative selection/background selection/ purifying selection) 是潜在的噪音
负选择会对正选择有一定的干扰作用,都能产生大量的低频突变,但是正选择会产生相对较多的高频突变。
两个亚群体之间的比较
多样性水平在亚群间比较,一般包括线性相关分析、亚群体间的差异比较两类。动植物重测序多是后者。Fst/pi ratio基于pi值。
- 群体分化程度Fst (Fixation index): 比较两个亚群体间的Pi值和亚群体内的Pi值的差异。
- 由PI值计算演变来(序列两两差异取均值)
- 两个亚群体在某一段seq区域的差异度。0是无差异,数值越大,则说明两个亚群体之间已经发生了明显的分化(亚群内个体相似,亚群间差异大)
1 | Fst=(\pi(between) - \pi(within))/ \pi(between) |
- 多样性变化倍数Pi ratio:某区间在亚群间的多样性差异的倍数,简单粗暴,就关注多样性值的高低变化。
- 例如野生群体A/栽培群体B;野生群体A的多样性较高,而栽培群体B的多样性较低,所以多样性降低最显著的基因组区域,就与驯化改良基因相关
- 其它比较值:ROD值、XP-CLR值等。而多个品种间的比较分化差异的di值
基于haplotype单体型的比较
前面pi/fst等都是基于SNP位点的多态性来检测潜在的选择信号区域。另一种方法是基于单体型haplotype的选择信号的检测。在selective sweeps选择过程中,有些强烈受到选择的位点variants由于LD的因素会连带着其附近的位点variants一起被保留,并且不会受到重组recombination的打断。一些低重组区域的haplotypes的长度会高于那些高重组区域的haplotypes的长度。因此,对比同一genomic区域在不同群体中的haplotype的长度可以用来判断是否受到选择。例如:在一个群体内部,如果某一个体强烈受到选择,其haplotype的长度会远长于其它个体;同理,对于两个群体之间的比较,某一群体受到选择,则其基因组中的受选择区域的haplotypes会比未受到选择群体中的haplotypes更长。
检测haplotype的选择信号最好利用定相phased后的数据集。方法有EHH和CLR法。这里利用R包中的rehh包进行分析。rehh有强大的说明和教程文档,后续深入了解其原理时值得进一步学习研究。rehh tutorial
rehh的实践
- 读取数据。分别读取两个群体的phased的vcf数据
polarize_vcf设为FALSE. because we have not used an outgroup genome to set our alleles as derived or ancestral- 根据maf进行过滤位点
1
2
3
4
5
6
7
8# read in data for each species# house
house_hh <- data2haplohh(hap_file = "./house_chr8.vcf.gz",polarize_vcf = FALSE)
# bactrianus
bac_hh <- data2haplohh(hap_file = "./bac_chr8.vcf.gz",polarize_vcf = FALSE)
## filter on MAF - 0.05
house_scan <- scan_hh(house_hh_f, polarized = FALSE)
bac_scan <- scan_hh(bac_hh_f, polarized = FALSE)

- 计算计算单体型的iHS值。
polarized = FALSEfreqbin =1if we know the ancestral allels or derived allels. rehh can apply weights to different bins of allele frequencies in order to test whether there is a significant deviation in the iHS statistic.- log Pvalue用于检测outliers值点,
1
2
3
4
5
6
7
8
9
10## perform haplotype genome scan- iHS
house_scan <- scan_hh(house_hh_f, polarized = FALSE)
bac_scan <- scan_hh(bac_hh_f, polarized = FALSE)
## perform iHS on house
house_ihs <- ihh2ihs(house_scan, freqbin = 1)
### plot the iHS statistics
ggplot(house_ihs$ihs, aes(POSITION, IHS)) + geom_point()
### plot the log P-value
ggplot(house_ihs$ihs, aes(POSITION, LOGPVALUE)) + geom_point()


- 计算群体之间的EHH值 xpEHH
- 计算 cross-population 的 EHH test。利用之前iHS检测出的iES值。
include_freq=T,we get the frequencies of alleles in our output, which might be useful if we want to see how selection is acting on a particular position1
2
3
4
5
6## perform xp-ehh
house_bac <- ies2xpehh(bac_scan, house_scan,
popname1 = "bactrianus", popname2 = "house",
include_freq = T)
#PLOT the xpEHH values
ggplot(house_bac, aes(POSITION, XPEHH_bactrianus_house)) + geom_point()
负数值代表在pop2(house in this case)中的强烈的选择信号。

- 检测受选择区域中的haplotype structure
1 | # find the highest hit 以最显著的位点做示例 |

house_furcation

bac_furcation
1 | # calculate the haplotype length around our signature of selection. |

house_furcation

bac_furcation
the blue haplotype is much larger around this target and is also more numerous in the European house sparrow.
- 输出数据
1 | # write out house bactrianus xpEHH |