Pophelper基本说明
群体遗传下游分析中的一项常规分析即是群体结构的分层展示(STRUCTURE ANALYSIS),不同于系统树和PCA,群体结构分层可以分断出小群体的个数,每个小群体之间的基因交流情况,甚至是小群体或者个体内的血源组成。
群体结构分层常用软件有STRUCTURE,ADMIXTURE和faststructure。STRUCTURE是群体结构分析的经典软件,但运行速度较慢。ADMIXTURE和faststructure软件等是近些年较新的软件,由于运算速度相对较快,已有了较多的引用次数。
群体结构分层的可视化展示通常是以堆叠柱状图所展示,Pophelper即是面向群体结构分层展示的强大的R包软件。
- 主页:[http://www.royfrancis.com/pophelper/]
- Web App(wrote by shiny) [https://roymf.shinyapps.io/structure/]
- 软件基本流程workflow:
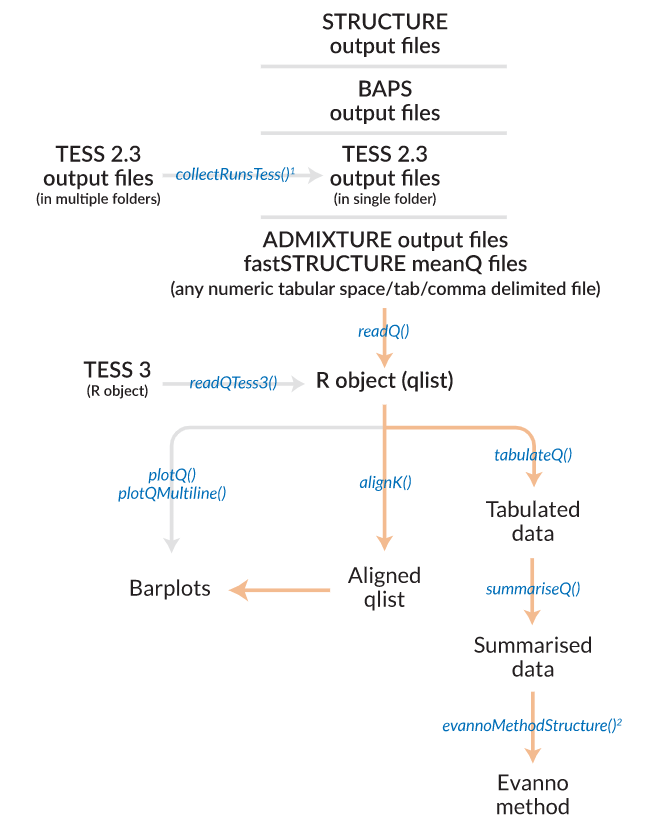
使用介绍
1. 安装
R version >3.5
1 | # install the dependency packages |
2. 读取文件
Pophelper接受structure,admixture,faststructure,tess等软件的输出文件。个人较熟悉的ADMIXTURE和faststructure,其输出文件结构都是以meanQ和meanP的矩阵文件。
1 | library(pophelper) |
3. 绘制最佳K值线
Pophelper中evannoMethodStructure()函数仅支持对STRUCTURE的结果绘制最佳K值线。其基本步骤包括三步
tabularQ(),接收读取的structure list文件summariseQ(),接收tabularQ返回结果evannoMethodStructure(),接收summ返回结果,绘制最佳K值线
1 | tbq <- tabulateQ(slist) |
- 最佳K值线结果
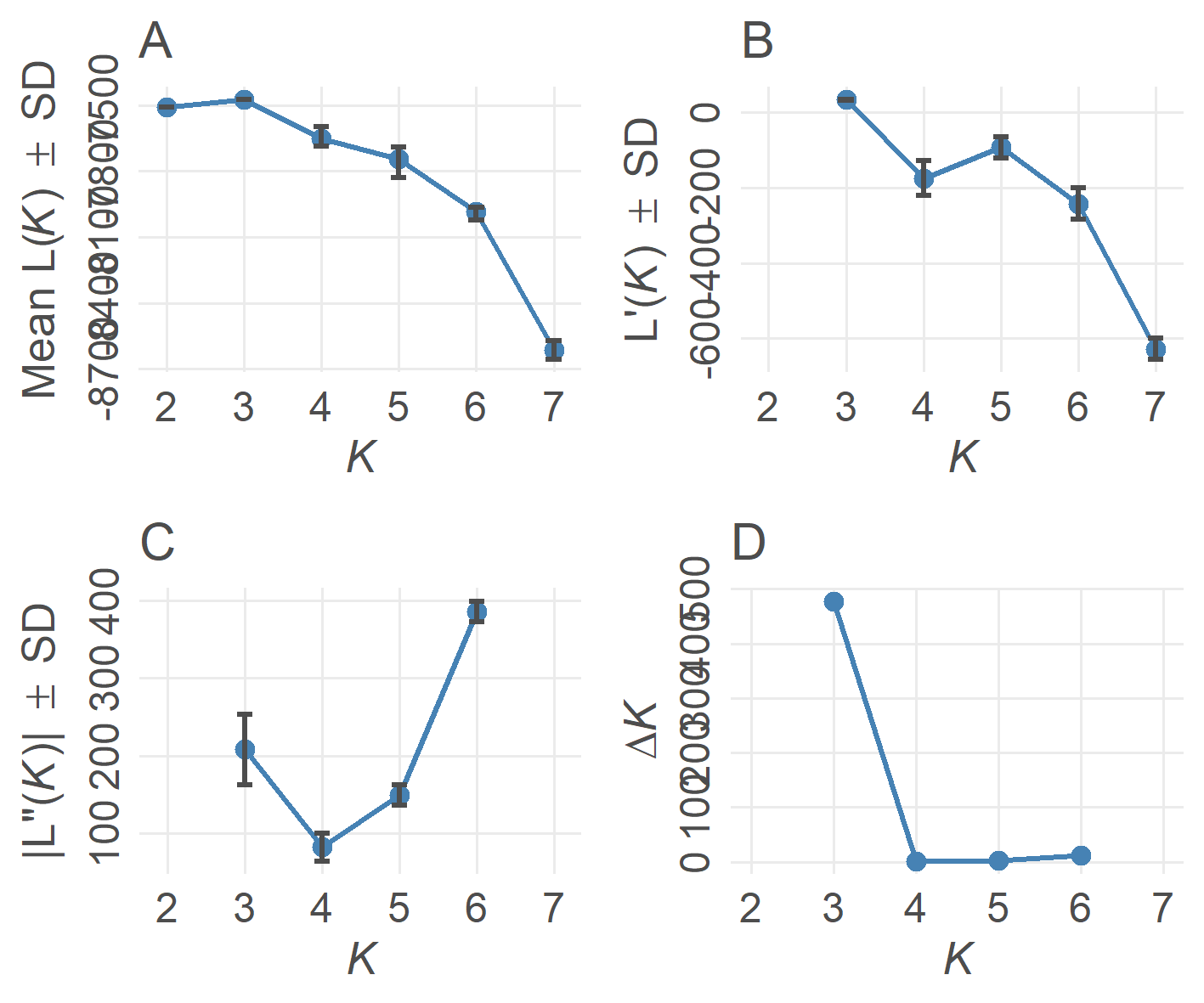
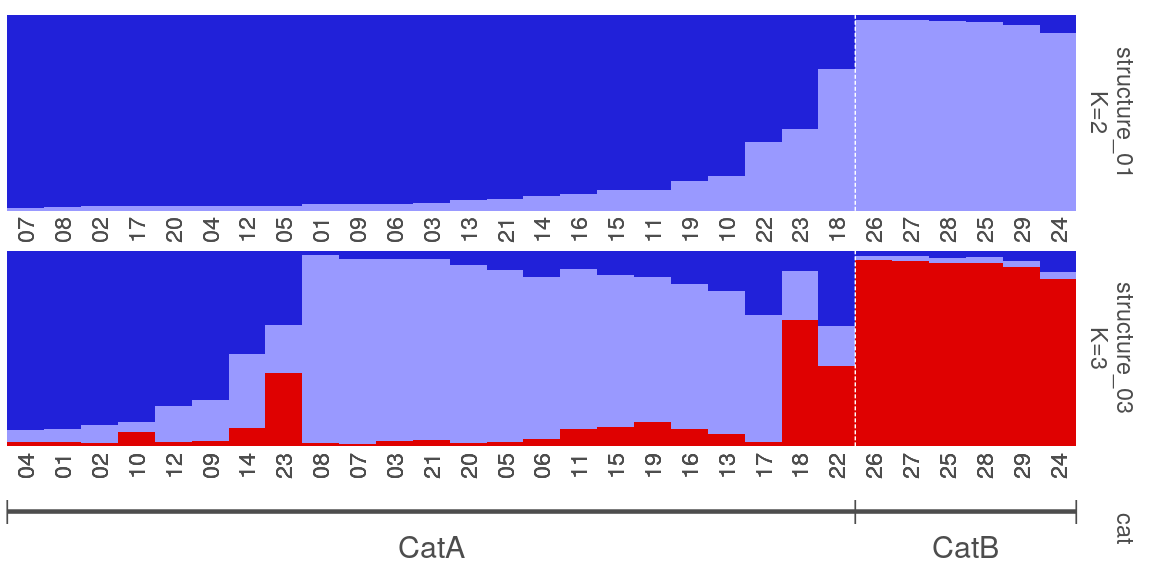
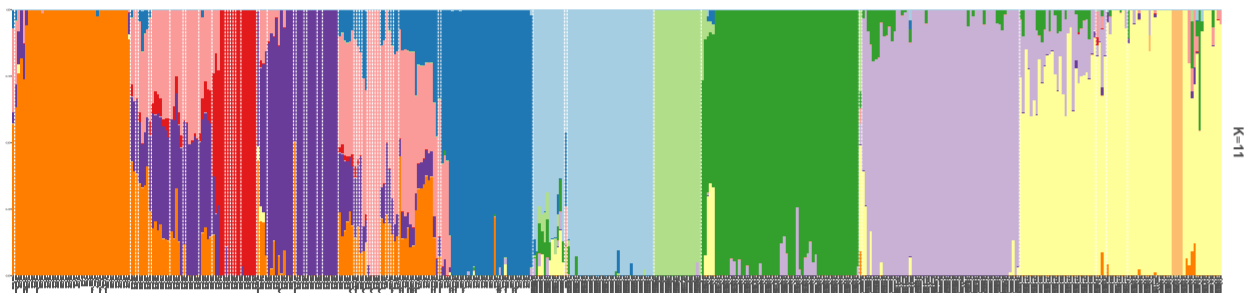
4. 绘制柱状堆叠图plotQ()
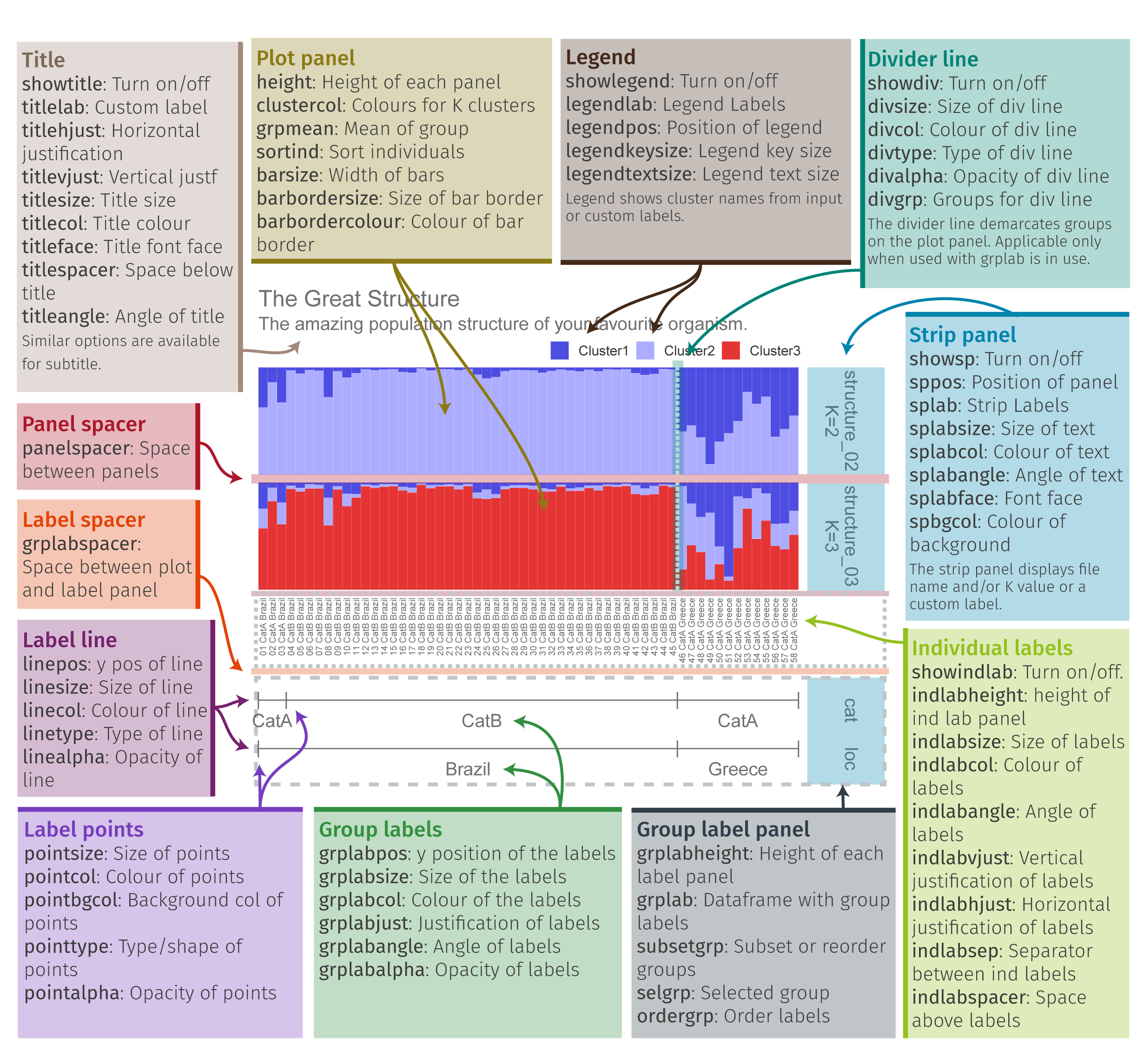
一个plotQ包含了复杂的柱状堆叠图的参数,一些常用参数:
imgoutput= "sep"/"join"默认sep,展示每个K值图,或者合并showsp=T:strip panel,展示每个K值堆叠图的标签sppos="left"splab="nameK1"splab=paste0("K=",sapply(slist[c(1,4:8)],ncol))仅显示K=num的标签spbgcol=..
clustercol=c("#A6CEE3", "#3F8EAA", "#79C360".....)堆叠柱状图的颜色showlegend=T:展示图例legend。useindlab=T:show individual lab 每个堆叠柱状图的label展示,需要q矩阵的rowname()。indlabsize,indlabcol
sortind="all"/"Cluster1"不设置时是默认是按照样本rowname()的顺序展示堆叠图,可设置cluster排序,或者个人手动调整matrix矩阵的样本顺序grplab=onelabset1(group label)在底部分组展示。含有group时设置sorted会同时显示。onelabeset1为列表,其顺序同是按照meanQ矩阵的rownames()顺序来的。grpsizegrpangle=90字体垂直展示
panel spacer=0.3对join合并图柱状堆叠图中间距离
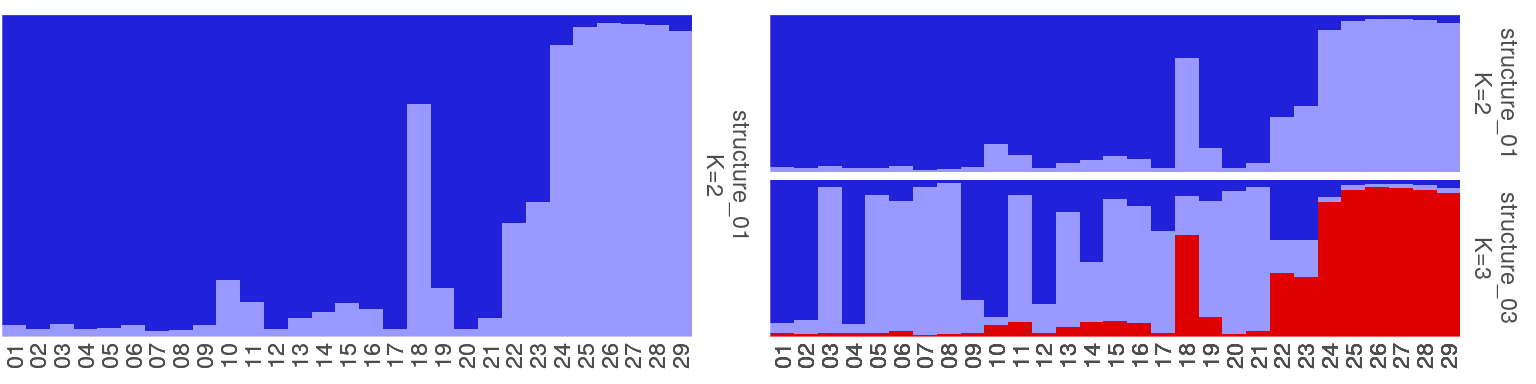
5. 一些群体结构图的示例
1 | sfiles <- list.files(path=system.file("files/structure",package="pophelper"), full.names=T) |
Reference
[http://www.royfrancis.com/pophelper/articles/index.html#plotq]